 Hekate
Hekate
XL-MS allows for the study of protein-protein binding by way of using small isotopically-labelled linker molecules. These linker molecules bind between residues that are within range of each other and then the resultant cross-linked protein complex is digested and analysed by mass spectrometry. The software developed by Andrew Holding to interrogate the mass spectrometry data was named Hekate and is freely available.
More details are available on the Hekate page of this site and the results of this work were published as:
- Holding et. al (2013) J. Proteome Res. [PubMed]
- Liu et. al (2013) Genes Dev. [PubMed]
- Rego et. al (2013) EMBO J. 32. 1334-1343. [Pubmed] [F1000Prime]
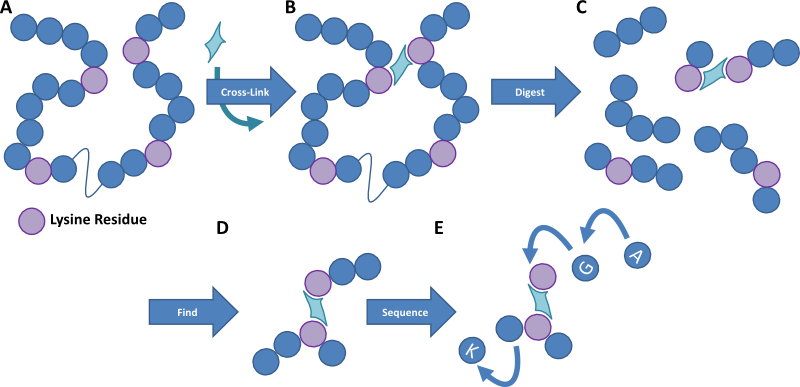
Figure 1. Cartoon image outlining the method of protein cross-linking. A protein sample is cross-linked with a homobifunctional reagent that links residues within a certain distance of each other. The protein sample is then digested into small peptides and the cross-linked fragments are detected by mass spectrometry. These peptides are then fragmented to provide an amino acid sequence detailing where in the protein the cross-linkers occur, and thereby we can work out the structure of the protein and how multiple proteins interact.
 Brundle
Brundle
Brundle is an R package that provides a series of functions for the normalisation of ChIP-Seq data to internal or external controls. It was developed to resolve challenges in estimating the precise fold-change between conditions.
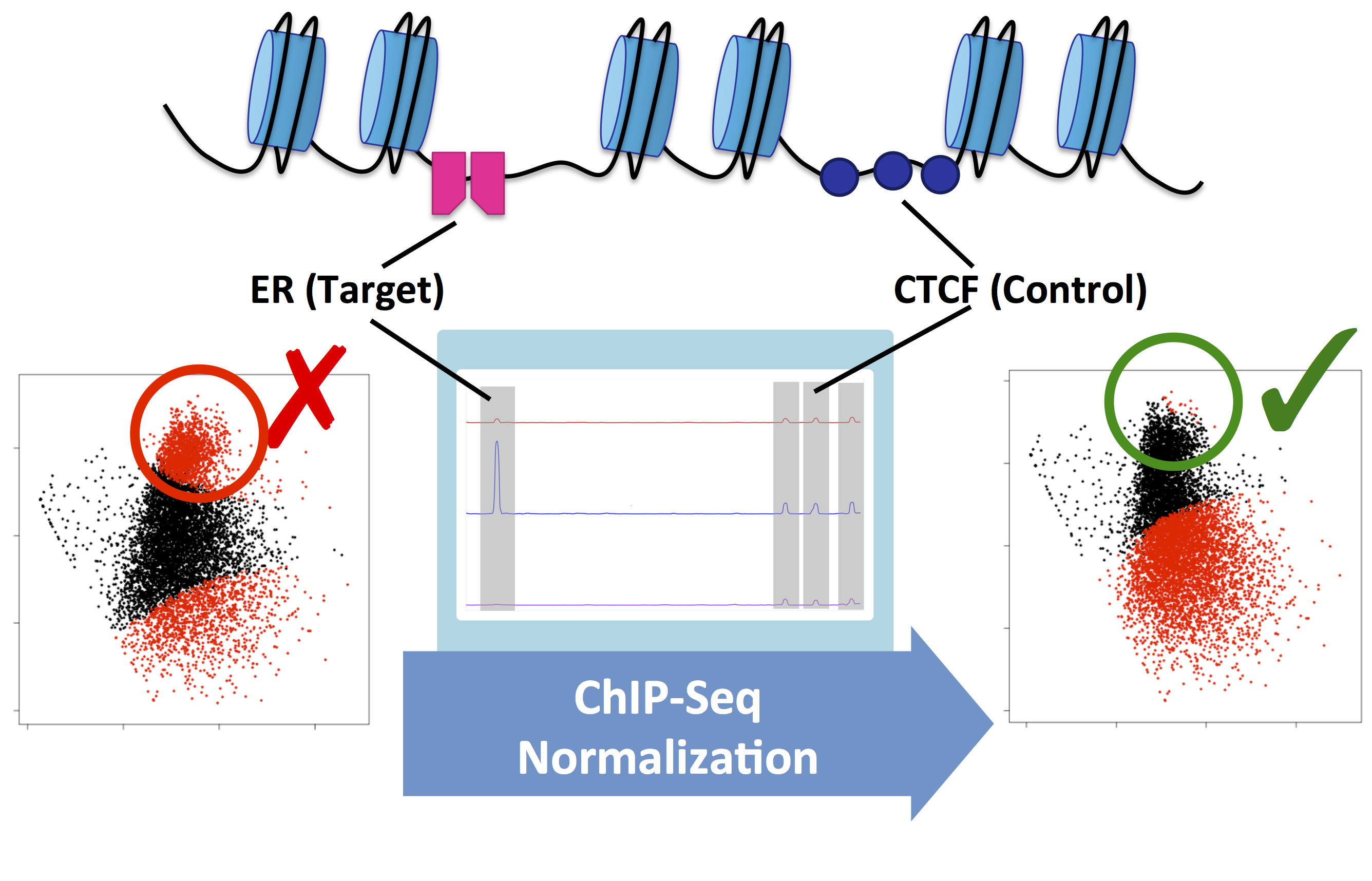
Figure 2. MA plots showing Estrogen Receptor-alpha (ER) ChIP-Seq data before and after the treatment of MCF7 cells with fulvestrant. This example shows the difference between the raw data (left) and normalised data (right). Without normalisation, biological signal can be supressed. In this example, normalisation was achieved using a second control antibody against CTCF. The CTCF binding sites were then used to normalise the ER binding data.
 VULCAN
VULCAN
VULCAN (VirtUaL ChIP-Seq Analysis through Networks) is a package that applies machine learning to patient data and uses the generated gene regulatory networks to infer co-factors that interact with the target factor in a ChIP-Seq experiment.
More details on the method and the results of this work were published as:
- Holding et. al (2019) Genome Bioloy. [bioRxiv]

Figure 3. 1. ChIP-Seq analysis from multiple conditions is undertaken to generate cistrome data at multiple timepoints (or conditions). Binding events are then compared using differential binding analysis to establish log-fold change values for individual binding events between each timepoint. 2. Network generation was undertaken with ARACNe-AP by inferring all pairwise TF-target coexpression from patient datasets (e.g. TCGA breast & METABRIC datasets). 3. All the targets of each specific TF in the network, i.e. the individual regulons, are tested against the established changes in ER binding through the msVIPER algorithm [15] to identify proteins that interact with the target transcriptional factor and final prediction is given for potential interacting co-factors.