Current research
Despite the overwhelming success of genetics to advance our understanding of cancer, it is still primarily a disease of signalling. The reason for this apparent contradiction is the critical lack of high-throughput methods to establish the topology of biological pathways.
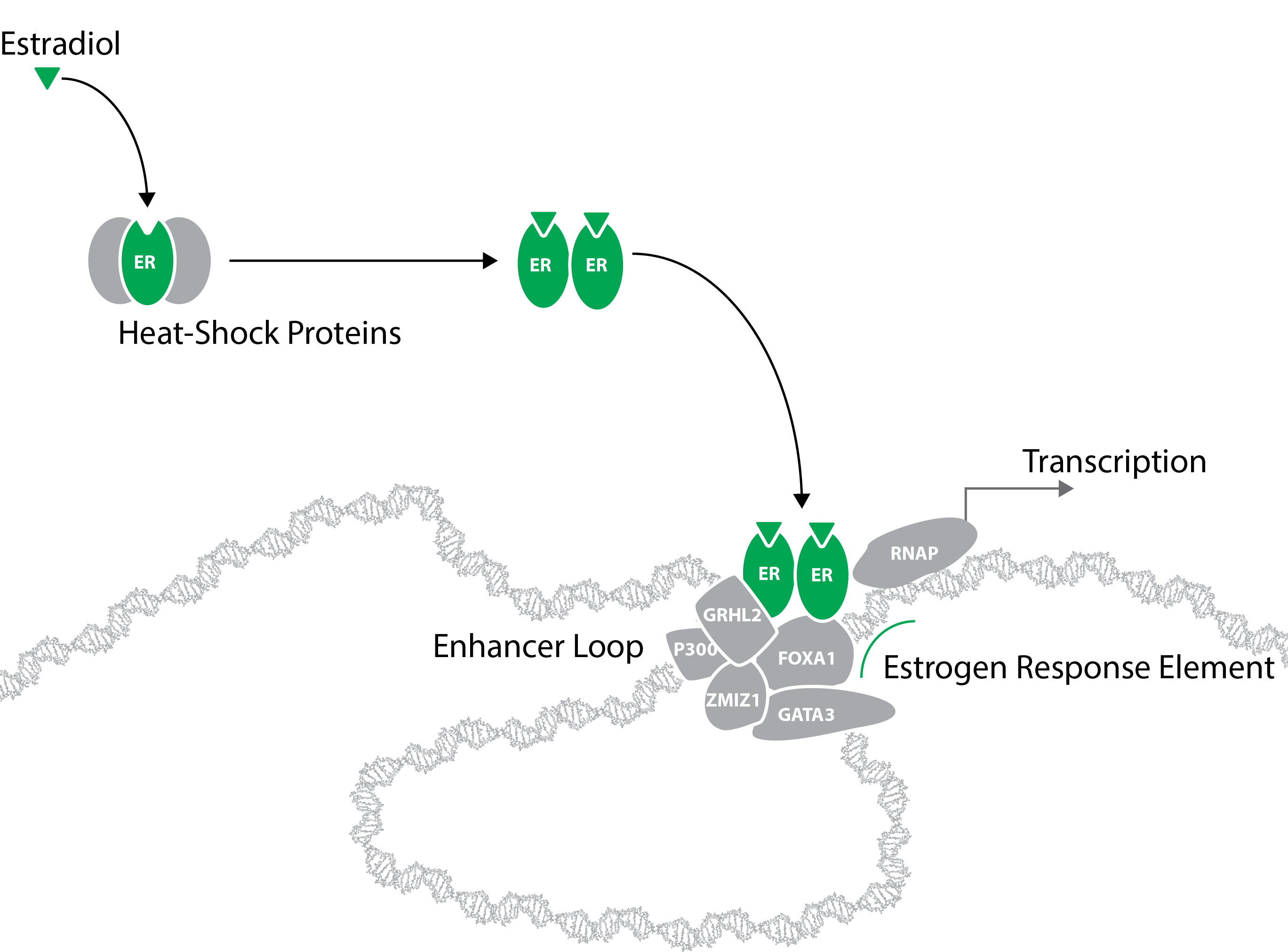
Andrew's research programme focuses on developing methods to meet this challenge and in applying them to the regulation of the estrogen receptor (ER) in breast cancer. In 70% of all breast cancer cases, this receptor drives the growth and proliferation of the tumour and is a key target for the drug Tamoxifen. Unfortunately for 10-40% of patients, depending on the stage of the disease at first diagnosis, the disease relapses and is often resistant to the most successful targeted therapies. As ER is known to associate with pioneer factors and numerous co-factors, including many with enzymatic activity that can modify the ER, a potential solution to resistance is to target them.
Andrew is applying the methods he developed to investigate the role of these co-factors and how they interact with the ER to produce a complete model of the signalling pathway, and to identify how the reprogramming of the pathway leads to resistance. The opportunities to undertake this kind of study are only recently possible and relies on state-of-the-art computational methods to combine proteomic and genomic data. In the long term, he aims to produce a model that could be used to predict the effects of perturbing components of the pathway with drugs and potentially lead to improved combinatorial treatment regimes.



